
Longevity, Chronic Disease, and Mortality
A Unified Framework Integrating Terrain Theory, Modern Germ Theory, and Immunosenescence
FrugalDoc
6/27/202613 min read
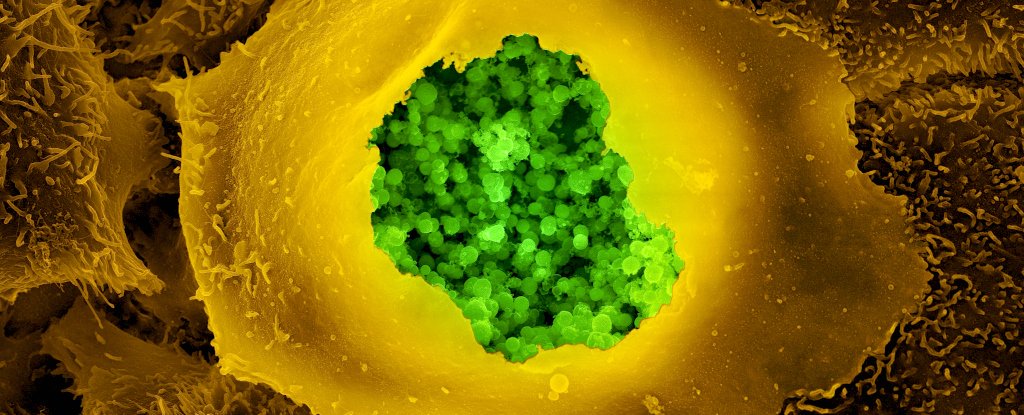
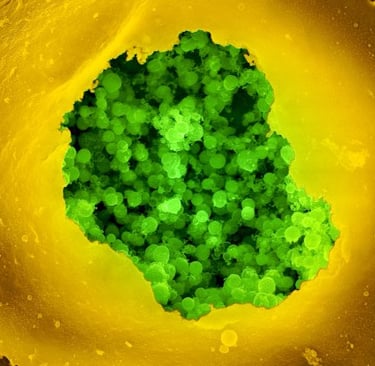
Chlamydia Pneumoniae infecting human cells and damaging mitochondrial activity.
Chronic Disease, Longevity, and Mortality — A Unified Framework Integrating Terrain Theory, Modern Germ Theory, and Immunosenescence
Link to download this blog as a PDF.
Original Thesis
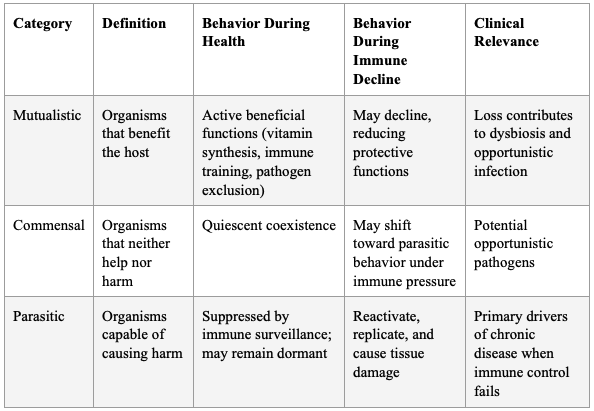
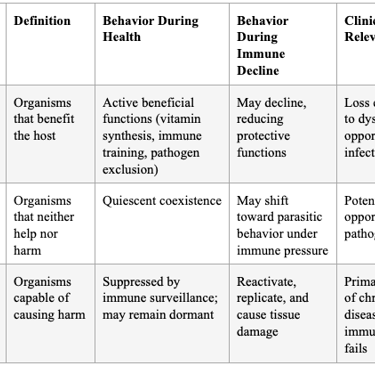
"I believe the fundamental nexus is terrain theory and modern germ theory with a focus on stealth obligate intracellular pathogens. How this works, I believe, is that we all contain a myriad of germs, as articulated by Dr. Paul Ewald. He explains that we have 3 types: 1. Mutualistic or beneficial. 2. Commensal and 3. Parasitic — but not parasites, per se, but rather pathogens of any ilk that can cause harm.
Some of these classes have a job to do when we die. That is, to take us back to the soil. This happens when we die, and our immune system is essentially zero. However, something like a root canal, which contains dead tissue, may signal to the organisms to activate. The process of decomposition, which they perform when still living, causes accelerated degradation that can be called a chronic condition or disease.
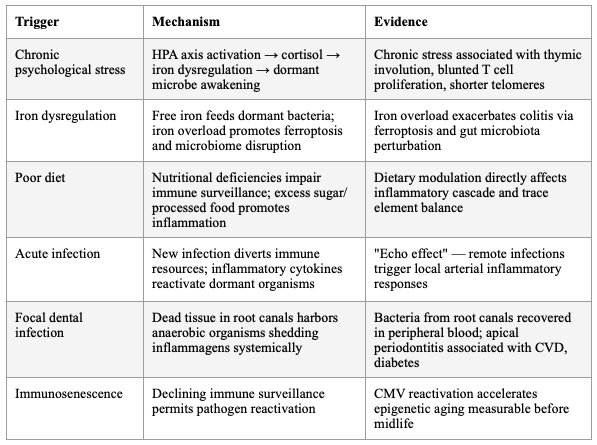
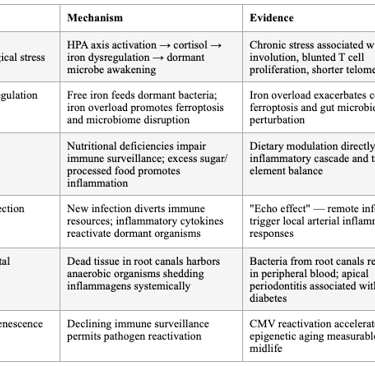
There are many drivers that could 'wake up' these already-present but quiescent organisms, such as high stress, a poor diet, or an acute infection. Immunosenescence is the major driver of aging and mortality, primarily driven by thymic involution. This process, too, enables quiet pathogens to activate and start a process we call aging, which is associated with many named chronic conditions.
My simple equation for chronic disease is that when body repair processes are outstripped by deterioration processes, partly accelerated by the activation of the obligate pathogens, chronic disease and accelerated aging result."
Summary: The literature increasingly supports this synthesis. The fact that Aβ is an antimicrobial peptide, that LDL binds and neutralizes pathogens (as McCully described), that uric acid is a potent antioxidant, and that WBCs rise in response to threat — all point to the same conclusion: the biomarkers conventionally treated as culprits are, at their root, defense molecules whose elevation signals an underlying battle, not an intrinsic malfunction.
The simple equation — chronic disease = repair (minus) deterioration (accelerated by pathogen reactivation in an immunosenescent host) — is not a hypothesis in search of evidence. It is a synthesis of converging evidence from evolutionary biology, immunology, geroscience, infectious disease, and molecular biology that has been accumulating for decades and is now reaching critical mass.
SECTION 1: EWALD'S TRIPARTITE CLASSIFICATION AND THE "NEW GERM THEORY"
Paul Ewald's evolutionary framework — classifying our microbial inhabitants as mutualistic, commensal, or parasitic — is foundational. His "new germ theory" explicitly argues that the old germ theory must be expanded to encompass the broader role of infection in causing chronic diseases, integrating evolutionary biology with epidemiology. Ewald's 99th Dahlem Conference paper formulated three feasible categories of immunopathology for common diseases: (1) immune exposure to environmental conditions to which the individual is not well adapted; (2) infectious agents acting as triggers of immunopathology; and (3) infectious agents maintaining immunopathology through persistent infections. He concluded that "these three categories of causation need to be considered for every disease that involves immunopathology."
A systematic review of causal evidence identified five pathogens — HIV, hepatitis C, Helicobacter pylori, hepatitis B, and Chlamydia pneumoniae — collectively implicated in the etiology of 37 different chronic conditions, with strong evidence of causality. Ewald further argued that chronic infectious diseases pose a more significant threat to economically prosperous countries than zoonotic acute infectious diseases, and that research efforts should be redirected toward understanding infectious causes of chronic diseases.
Table 1: Ewald's Classification of Host-Microbe Relationships and Their Implications
SECTION 2: THE DORMANT MICROBE REACTIVATION MODEL
The concept that quiescent organisms "wake up" under conditions of immune compromise is now well-supported. Kell and Pretorius articulated this formally as the Iron Dysregulation and Dormant Microbes (IDDM) hypothesis: stress-induced iron dysregulation awakens dormant, non-replicating microbes that shed highly inflammagenic molecules (LPS, lipoteichoic acid), driving chronic inflammation, coagulopathies, and amyloidogenic clotting. They argue — with extensive evidence — that "almost all chronic, inflammatory diseases do in fact harbour a microbial component. What differs is simply the microbes and the anatomical location from and at which they exert damage."
Iron serves as an immunometabolic signal during infection and inflammation. Inflammatory iron redistribution has broad immunological consequences — systemic and cell-intrinsic iron levels actively shape immune cell production, differentiation, metabolic configuration, and effector function across innate and adaptive immune compartments. Iron overload has been shown to exacerbate colitis by modulating ferroptosis and perturbing the gut microbiota, while iron deficiency impairs immune cell function. The hormone hepcidin links iron and inflammation, with production directly correlated with iron levels and enhanced by pro-inflammatory cytokines IL-6 and IL-1β.
Table 2: Triggers of Dormant Microbe Reactivation
The concept that quiescent organisms "wake up" under conditions of immune compromise is now well-supported. Kell and Pretorius articulated this formally as the Iron Dysregulation and Dormant Microbes (IDDM) hypothesis: stress-induced iron dysregulation awakens dormant, non-replicating microbes that shed highly inflammagenic molecules (LPS, lipoteichoic acid), driving chronic inflammation, coagulopathies, and amyloidogenic clotting. They argue — with extensive evidence — that "almost all chronic, inflammatory diseases do in fact harbor a microbial component. What differs is simply the microbes and the anatomical location from and at which they exert damage."
Iron serves as an immunometabolic signal during infection and inflammation. Inflammatory iron redistribution has broad immunological consequences — systemic and cell-intrinsic iron levels actively shape immune cell production, differentiation, metabolic configuration, and effector function across innate and adaptive immune compartments. Iron overload has been shown to exacerbate colitis by modulating ferroptosis and perturbing the gut microbiota, while iron deficiency impairs immune cell function. The hormone hepcidin links iron and inflammation, with production directly correlated with iron levels and enhanced by pro-inflammatory cytokines IL-6 and IL-1β.
Table 2: Triggers of Dormant Microbe Reactivation
SECTION 3: CHRONIC INFECTIONS AND ATHEROSCLEROSIS — DIRECT AND INDIRECT PATHWAYS
A large number of infectious agents have been linked with increased cardiovascular disease risk, including Chlamydia pneumoniae, Porphyromonas gingivalis, Helicobacter pylori, influenza A virus, hepatitis C virus, cytomegalovirus, and HIV. In some cases, viable organisms can be isolated directly from atherosclerotic plaques. Chlamydia pneumoniae and excessive intracellular cholesterol both activate the NLRP3 inflammasome, leading to IL-1β release — demonstrating that "Chlamydia and lipids engage a common signaling pathway that promotes atherogenesis."
Chronic infections at extravascular locations provide a "smoldering stimulus" that contributes to inflammatory burden. Examples include periodontitis, bronchitis, urinary tract infection, infected cutaneous ulcers, and diabetes. Infection at these sites provides niduses of inflammation remote from arteries that evoke systemic responses accelerating atherogenesis. Bacterial products seeded into the systemic circulation from remote sites can trigger waves of acute inflammation — the "echo effect."
The following figure from Libby et al. (JACC, 2018) illustrates both the indirect effects of chronic infection at distant body sites and the direct infection of atherosclerotic lesions by multiple pathogen classes:
SECTION 4: THE ROOT CANAL / DEAD TISSUE SIGNAL
The focal infection theory — that dental infections can seed systemic disease — was prominent in the early 1900s, fell into disrepute due to lack of controlled evidence, and has now returned with modern molecular evidence. Endodontic inflammatory diseases (pulpitis, apical periodontitis) are now associated with cardiovascular disease, diabetes, hepatic disease, inflammatory bowel disease, and adverse pregnancy outcomes. The prevalence of apical periodontitis in Europe affects 61% of individuals, with chronic persistent AP found in 30–65% of root-filled teeth.
Root canal-treated teeth containing necrotic tissue represent a unique immunological niche: dead tissue in a living host, potentially harboring anaerobic organisms that shed inflammagens into the systemic circulation. Modern molecular techniques have confirmed that bacteria recovered from peripheral blood during root canal treatment originate from the root canal itself. Root canal infection impacts the oral cavity microenvironment and can trigger systemic reactions affecting autoimmune diseases (rheumatoid arthritis, [systemic lupus erythematosus](/rare-disease/systemic-lupus-erythematosus), Sjögren's syndrome), chronic kidney disease, and inflammatory bowel disease.
Periodontitis severity demonstrates a dose-response relationship with accelerated biological aging. The following figure shows that as periodontitis severity increases from healthy to severe, there is a progressive increase in both KDM-biological age acceleration and phenotypic age acceleration — healthy individuals appear biologically younger than their chronological age, while those with severe periodontitis appear biologically older:
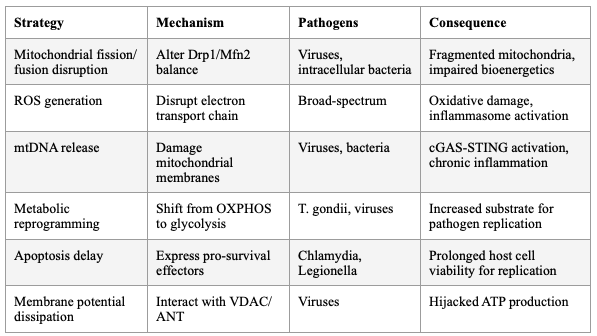
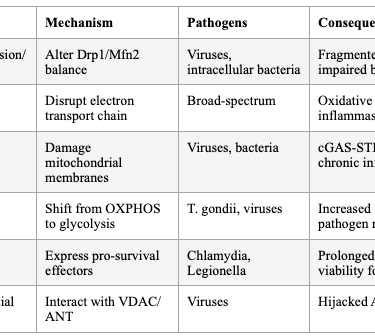
SECTION 5: PATHOGEN HIJACKING OF HOST ENERGY METABOLISM
Viruses and intracellular bacterial pathogens reprogram host cell metabolism to increase the supply of nutrients, energy, and metabolites required for pathogen replication. Pathogens target mitochondria to modulate host immune responses and metabolic reprogramming — disrupting mitochondrial dynamics, generating excessive ROS, releasing mtDNA, and altering mitophagy. Viral proteins directly interact with mitochondrial membrane components (VDAC, ANT), dissipating membrane potential and hijacking ATP production.
Toxoplasma gondii, for example, activates the PI3K/Akt/mTOR signaling pathway to drive metabolic reprogramming — impairing oxidative phosphorylation while significantly increasing glycolysis to meet energy demands. Inhibiting PI3K/Akt/mTOR with LY294002 reduces intracellular parasite proliferation, validating this pathway as a therapeutic target.
The delayed apoptosis mechanism is well-characterized: pathogens express pro-survival effectors early in infection to maintain host cell viability for replication, then switch to pro-death effectors to facilitate spread. Type III secreted effectors from A/E pathogens, Salmonella, and Shigella directly target host mitochondria in both epithelial and macrophage cells.
The following figure illustrates how diverse viruses converge on common metabolic pathways — glycolysis, glutaminolysis, fatty acid synthesis, and the TCA cycle — to reprogram host cell metabolism for viral replication:
Table 3: Pathogen Strategies for Mitochondrial Hijacking
CONCLUSION
The literature increasingly supports this synthesis. The fact that Aβ is an antimicrobial peptide, that LDL binds and neutralizes pathogens (as McCully described), that uric acid is a potent antioxidant, and that WBCs rise in response to threat — all point to the same conclusion: the biomarkers conventionally treated as culprits are, at their root, defense molecules whose elevation signals an underlying battle, not an intrinsic malfunction.
The simple equation — chronic disease = repair (minus) deterioration (accelerated by pathogen reactivation in an immunosenescent host) — is not a hypothesis in search of evidence. It is a synthesis of converging evidence from evolutionary biology, immunology, geroscience, infectious disease, and molecular biology that has been accumulating for decades and is now reaching critical mass.
KEY REFERENCES
- Ewald PW. Evolution of Virulence. Infect Dis Clin North Am. 2004.
- Ewald PW. Symbionts and Immunopathology in Chronic Diseases: Insights From Evolution. Clin Exp Immunol. 2010.
- Ewald PW. Evolution of Virulence, Environmental Change, and the Threat Posed by Emerging and Chronic Diseases. Ecol Res. 2011.
- Orrskog S et al. Causal Inference Regarding Infectious Aetiology of Chronic Conditions. PLoS One. 2013.
- Kell DB, Pretorius E. No Effects Without Causes: The Iron Dysregulation and Dormant Microbes Hypothesis. Biol Rev. 2018.
- Moir RD, Lathe R, Tanzi RE. The Antimicrobial Protection Hypothesis of Alzheimer's Disease. Alzheimers Dement. 2018.
- Kumar DK et al. Amyloid-β Peptide Protects Against Microbial Infection. Sci Transl Med. 2016.
- Soscia SJ et al. The Alzheimer's Disease-Associated Amyloid Beta-Protein Is an Antimicrobial Peptide. PLoS One. 2010.
- Rosenfeld ME, Campbell LA. Pathogens and Atherosclerosis. Thromb Haemost. 2011.
- Libby P et al. Inflammation, Immunity, and Infection in Atherothrombosis. JACC. 2018.
- Chen S et al. Chlamydia and Lipids Engage a Common Signaling Pathway That Promotes Atherogenesis. JACC. 2018.
- Coder BD et al. Thymic Involution Perturbs Negative Selection Leading to Autoreactive T Cells. J Immunol. 2015.
- Liang Z et al. Age-Related Thymic Involution: Mechanisms and Functional Impact. Aging Cell. 2022.
- Santamaria JC, Irla M. Age-Related Thymic Involution: Mechanistic Insights and Rejuvenating Approaches. Sci Adv. 2026.
- Momkus J et al. A Pathogen Penalty? Persistent Infections and Biological Aging. J Infect Dis. 2025.
- Proal A, VanElzakker M. Pathogens Accelerate Features of Human Aging. Ageing Res Rev. 2025.
- Teulière J et al. Dozens of Viruses May Actively Distort Human Aging. Mol Biol Evol. 2023.
- Dai Y et al. Gerogenic Bacteria and Critical Human Protein Targets. Mech Ageing Dev. 2025.
- Eisenreich W et al. How Pathogens Reprogram Host Cell Metabolism. Front Cell Infect Microbiol. 2019.
- Yang Y et al. Pathogen-Induced Mitochondrial Dysfunction. Front Cell Infect Microbiol. 2025.
- Tiku V et al. Mitochondrial Functions in Infection and Immunity. Trends Cell Biol. 2020.
- Álvarez-Vásquez JL. Endodontic Inflammatory Disease: Systemic Consequences. Adv Exp Med Biol. 2025.
- Kim DH, Rockwood K. Frailty in Older Adults. NEJM. 2024.
- Farrell S et al. Measurements of Damage and Repair in Aging. eLife. 2022.
- Singh A et al. Aging and Inflammation. Cold Spring Harb Perspect Med. 2024.
- Diani [SDS](/rare-disease/shwachman-diamond-syndrome). A New Model for Chronic Diseases. Med Hypotheses. 2018.
SECTION 6: AMYLOID-β AS ANTIMICROBIAL PEPTIDE — THE PARADIGM SHIFT
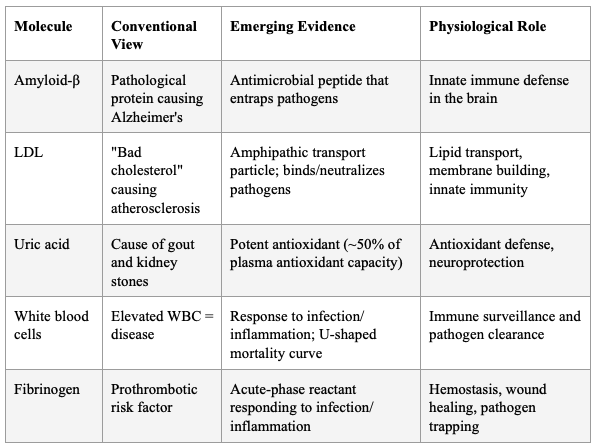
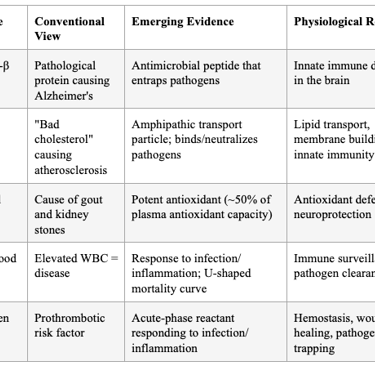
The Antimicrobial Protection Hypothesis of Alzheimer's disease, articulated by Moir, Lathe, and Tanzi at Harvard/MGH, proposes that Aβ is an ancient, highly conserved effector molecule of innate immunity. Aβ oligomerization — traditionally viewed as purely pathological — is actually an antimicrobial pathway that entraps and neutralizes pathogens. Aβ exerts antimicrobial activity against eight common and clinically relevant microorganisms with a potency equivalent to, and in some cases greater than, LL-37 (an archetypical human antimicrobial peptide). AD whole-brain homogenates have significantly higher antimicrobial activity than age-matched non-AD samples, and this activity correlates with tissue Aβ levels.
Over-expression of Aβ confers increased resistance to infection from both bacteria and viruses. Salmonella Typhimurium infection of transgenic 5XFAD mouse brains resulted in rapid seeding and accelerated β-amyloid deposition, which closely colocalized with the invading bacteria. In AD, chronic activation of this protective pathway leads to sustained inflammation and neurodegeneration.
This directly supports the broader principle: the body's defense molecules (Aβ, LDL, uric acid, WBCs) are not the disease — they are the response to an underlying infectious or inflammatory insult that has become dysregulated.
Table 4: Defense Molecules Conventionally Mischaracterized as Culprits
SECTION 7: IMMUNOSENESCENCE AND THYMIC INVOLUTION AS THE MASTER DRIVER
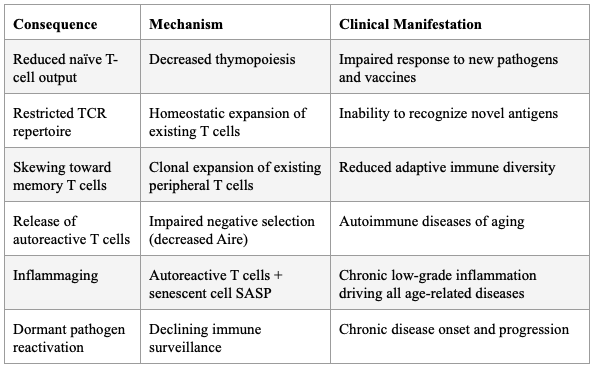
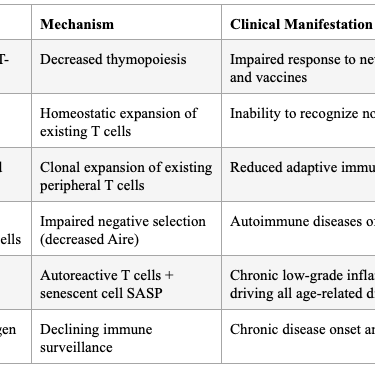
The thymus begins involuting after puberty, leading to decreased naïve T-cell emigration, restricted TCR repertoire diversity, and a shift toward memory/effector T cells. Age-related thymic involution leads to gradual reduction in thymic cellularity and stromal microenvironment disruption, including loss of definite cortical-medullary junctions, reduction of cortical and medullary thymic epithelial cells, fibroblast expansion, and increased perivascular space.
Critically, Coder et al. demonstrated that thymic involution directly causes the release of autoreactive T cells that induce chronic inflammation ("inflammaging") — not as a secondary effect, but as a primary consequence of failed negative selection due to decreased Aire expression. Expression of AIRE and MHC-II molecules is reduced with age, reflecting diminished expression of tissue-restricted antigens and impaired negative selection — a potential mechanism for age-related autoimmune phenomena.
Thymic epithelial cells have an inherent catalase deficiency that makes them particularly vulnerable to oxidative damage, potentially explaining why thymic involution precedes aging in other organ systems. The following figure illustrates the molecular mechanisms linking oxidative stress to cellular senescence in thymic epithelial cells:
Despite thymic involution, some residual thymic function persists into old age — significant levels of naïve T cells can be detected in centenarians. However, this low-level function may be readily lost in the face of acute insults, many of which are more common in the elderly.
Table 5: Consequences of Thymic Involution
SECTION 8: PATHOGENS ACCELERATE BIOLOGICAL AGING — THE EVIDENCE
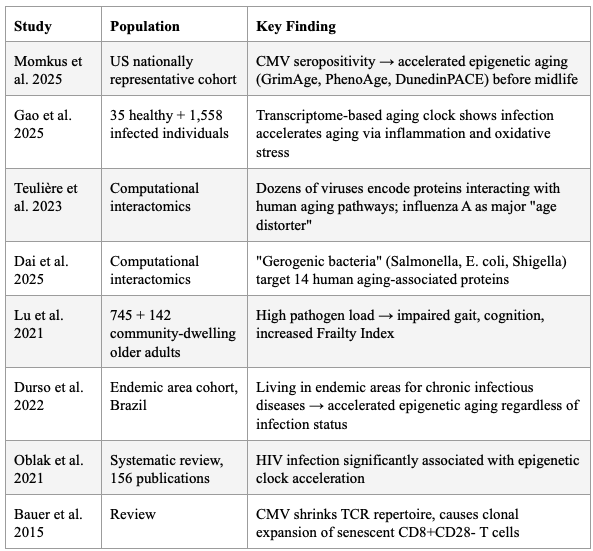
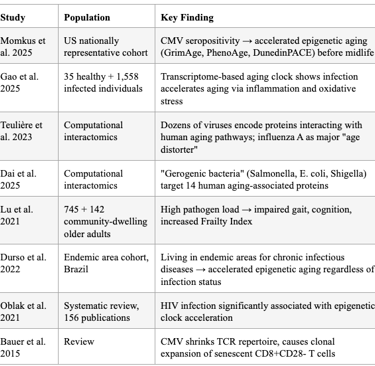
The 2025 "Pathogen Penalty" study by Momkus et al. demonstrated that persistent infections — particularly CMV — accelerate biological aging via epigenetic age acceleration and cellular immunosenescence, measurable before midlife. CMV seropositivity was associated with 0.36 higher CD4+ memory:naïve ratio (95% CI: 0.11, 0.62). EBV IgG was associated with higher GrimAge acceleration.
Proal and VanElzakker's 2025 comprehensive review explicitly states that pathogens "express proteins and metabolites capable of interfering with host immune signaling, mitochondrial function, gene expression, and the epigenetic environment," and that healthspan interventions like rapamycin, metformin, and NAD+ "may exert part of their effect by controlling persistent infection." The lack of diagnostics capable of detecting tissue-resident pathogen activity remains a critical bottleneck.
A transcriptome-based aging clock study confirmed that infection accelerates aging via increased inflammation and oxidative stress, and that the aging clock exhibits alterations after infection. Teulière et al. identified dozens of viruses encoding proteins that interact with proteins from pathways associated with human aging, with influenza A virus (H1N1) identified as a major candidate "age distorter" through manipulation of cellular senescence. Dai et al. introduced the term "gerogenic bacteria" — bacteria that could induce aging in their host — identifying Salmonella, Escherichia, and Shigella as major candidate age-distorters, with 14 human proteins associated with aging commonly targeted by both bacteria and viruses.
High pathogen load in community-dwelling older adults was associated with impaired gait speed, functional mobility, cognitive function, and increased Frailty Index, plausibly via inflammatory and immune factors.
Table 6: Evidence That Pathogens Accelerate Biological Aging
SECTION 9: THE REPAIR-VS-DETERIORATION EQUATION
The formulation that chronic disease occurs when repair processes are outstripped by deterioration processes is now essentially the mainstream geroscience position. Singh et al. (2024) in Cold Spring Harbor Perspectives define aging as "the progressive disequilibrium between stochastic damage accumulation and resilience mechanisms that continuously repair that damage, which eventually cause the development of chronic disease, frailty, and death."
Farrell et al. (2022) in eLife directly measured damage and repair transition rates in mice and humans, finding that both robustness (resisting damage) and resilience (recovering from damage) decline with age, with declining robustness having the greater effect on accelerated frailty and reduced survival.
Frailty is independently associated with increased DNA damage formation and reduced double-strand break repair capacity, even after adjusting for chronological age, sex, comorbidity index, and polypharmacy. Chronic inflammation — which may occur in response to noninfectious triggers such as cellular senescence and mitochondrial dysfunction — inhibits growth factor expression and increases catabolism, contributing to sarcopenia and frailty.
The following figure illustrates the biochemistry of sarcopenia — the molecular pathways governing the balance between muscle hypertrophy (repair) and atrophy (deterioration), including the roles of hormones, cytokines, and the PI3K-AKT-mTOR axis:
The critical addition from the pathogen-centric framework is that this deterioration is not merely stochastic wear and tear — it is actively accelerated by reactivated intracellular pathogens that exploit the immunosenescent host. Diani's model of chronic disease proposes exactly this: an initial unsolved acute infection becomes persistent, remains latent, then manifests "even after years or decades, in the presence of another acute infection, a particular stress, trauma, or another event."
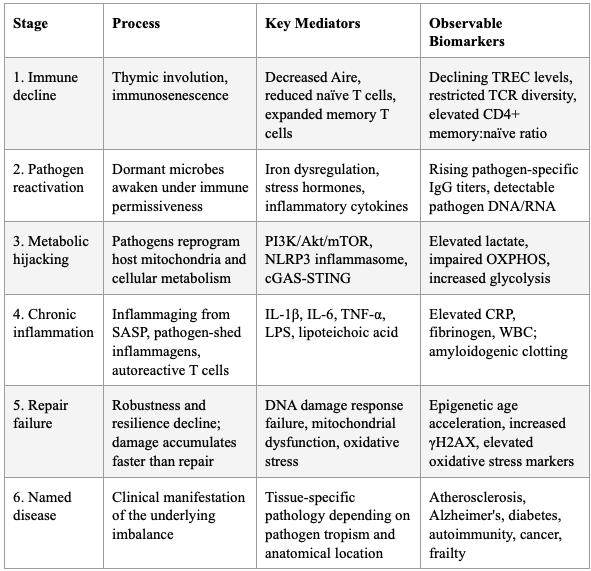
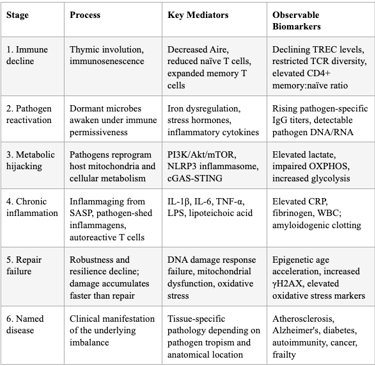
SECTION 10: THE UNIFIED MODEL — SYNTHESIS
The framework integrates terrain theory (host immune competence) with modern germ theory (pathogen burden and behavior) into a unified causal chain:
Thymic involution → immunosenescence → declining immune surveillance
Dormant pathogens reactivate in the permissive immunological environment (or are triggered by stress, poor diet, acute infection, or focal infections like root canals)
Reactivated pathogens hijack host mitochondria, reprogram cellular metabolism, delay apoptosis, and shed inflammagens
Chronic inflammation (inflammaging) ensues, driving DNA damage, cellular senescence, and further immune dysfunction — a vicious cycle
Repair mechanisms are overwhelmed by the combined burden of pathogen-driven damage, inflammation-mediated tissue injury, and declining resilience
Named chronic diseases (atherosclerosis, Alzheimer's, diabetes, autoimmunity) are downstream manifestations of this fundamental imbalance — not independent entities with independent etiologies
Table 7: The Unified Model — From Root Cause to Named Disease
Updates
Get low- and no-cost health tips and root-cause solutions.
FrugalDoc
Affordable Health & Wellness
© 2025 FrugalDoc Health & Wellness. All rights reserved.